The promiscuity ability of cowpea enables it to form nitrogen-fixing root nodules with diverse symbiotic bacteria. It is mainly nodulated by slow-growing bacteria which constitute heterogeneous group of rhizobia called “cowpea miscellany” belonging to the genus Bradyrhizobium. Currently the genus Bradyrhizobium comprises forty-one type strains ( http://www.bacterio.cict.fr/) while type strains isolated from other hosts such as Bradyrhizobium daqingense, Bradyrhizobium huanghuaihaiense, Bradyrhizobium paxllaeri, Bradyrhizobium ottawaense, Bradyrhizobium yuanmingense are also capable of nodulating cowpea. In order to gain insight into the genetic diversity of indigenous rhizobia isolated from nodule trapped from soil collected from three different geographical regions (Niger, Kaduna, Kano) in Nigeria, a detailed multilocus sequence analysis of concatemers of two protein-coding genes (glnll-recA) was performed.
The DNA fragments of the following loci were amplified with their respective primers using pure single colony of rhizobia strains diluted in sterile water and amplified by polymerase chain reaction with primers TSglnllF&TSglnllR for glnll-gluthamine synthetase ll and TSrecAF&TSrecAR for recA-DNA recombination protein. The partial gene sequences obtained together with sequences retrieved from GenBank were aligned using the CLUSTAL W software in the MEGA 7.0 software package. Phylogenetic trees were constructed using the neighbour-joining (NJ) methods in MEGA 7.0 software package. The gene sequences were concatenated using R software.
|
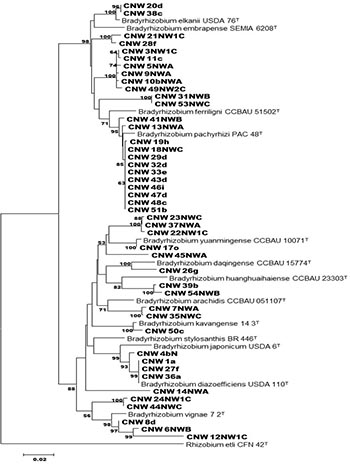
Bradyrhizobium species are divided into well-supported phylogenetic lineages designated I (represented by Bradyrhizobium japonicum) and II (represented by Bradyrhizobium elkanii) based on phylogenetic analyses of rrs gene and 16S-23S rRNA IGS. All strains included in this study clearly seperate into the two different groups as shown in Figure 1. The phylogenetic analysis based on the concatenated sequences is more robust and confident. Within the Bradyrhizobium elkanii group, strains CNW 20d, CNW 38c clustered with Bradyrhizobium elkanii, while ten of the strains clustered with Bradyrhizobium pachyrhizi. Within the Bradyrhizobium japonicum group, CNW 17o clustered with Bradyrhizobium yuanmingense, CNW 4bN, CNW 1a, CNW 27f, CNW 36a clustered with Bradyrhizobium diazoefficiens, CNW 50c clustered with Bradyrhizobium kavangense. Based on pairwise comparisons of the two concatenated sequences, the isolates CNW 4bN, CNW 1a, CNW 27f, CNW 36a displayed sequence similarities to USDA 110T, 99.1%, 100%, 100%, 100% respectively. CNW 20d, CNW 38c showed sequence similarity with to USDA 76T, 99.5%, 99.5% respectively. All the strains that clustered with PAC 48T showed sequence similarity of 99.1%. |
The close phylogenetic relationships with strains used as inoculants render them worthy for further investigation as inoculants in fields with similar edapho-climatic conditions. Therefore, studies investigating indigenous rhizobia in fields without rhizobia inoculation history are of importance for selecting novel strains adapted to the local environmental conditions. Future studies on symbiotic properties are needed to demonstrate whether they represent novel symbiovars.
Ojo Comfort Tinuade, Wageningen University & Research, The Netherlands (Click here for her 2017 update)